3 Vital Things You Should Know About Change Control in Pharmaceutical Projects to Run Them Smoothly
Understand What Exactly Change Control Means
Understand the Regulatory Background
Understand the Best Practices for Change Control
If you are involved in the change control process at your company, or if you are a decision-maker who wants to improve your existing systems, and work in the quality control, quality assurance, or regulatory affairs, you must understand the new aspects and best practices of Change control.
A change control system is a crucial part of the quality management system (QMS) of every company. Any change that is announced or requested must be cautiously checked, fully documented, and authorized.

A robust change control system will help manage changes of all types. It will help avoid inappropriate changes from happening. The Quality function and any other team that might be impacted by the change must conduct a complete review of all GMP-appropriate changes.
The change control process will run smoothly only if all the departments involved in the process are working in tandem and are aware of what needs to be considered. They must understand all the appropriate aspects of the complete change control process and the impact a change might have.
Annex 15 of the EU GMP Guidelines defines "change control" as:
"A formal system by which qualified representatives of appropriate disciplines review proposed or actual changes that might affect the validated status of facilities, systems, equipment or processes. The intent is to determine the need for action that would ensure and document that the system is maintained in a validated state."
Chapter 5.25 of the EU GMP Guidelines states the following about handling of changes.
"Significant amendments to the manufacturing process, including any change in equipment or materials, which may affect product quality and/or the reproducibility of the process should be validated."
ICH Q10
A model for an effective QMS is described in ICH Q10 which includes a section on change management. The following table from ICH Q10 highlights application of process performance and product quality monitoring system throughout the product lifecycle.
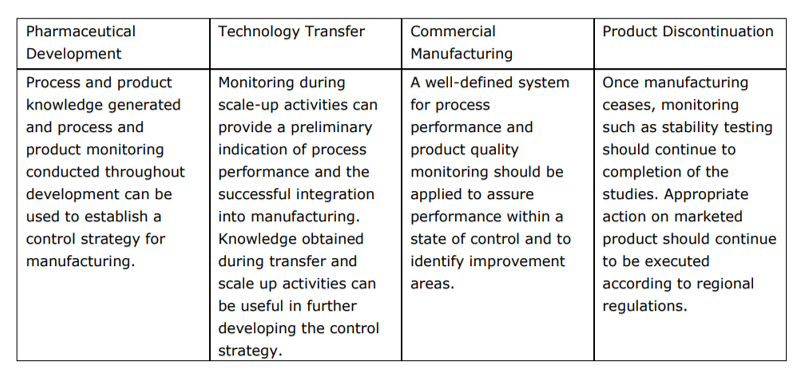
The Code of Federal Regulations (CFR) states the following about "change control" (21 CFR, 211.100 and 21 CFR, 211.160):
§ 211.100 Written procedures; deviations.
(a) "There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess. Such procedures shall include all requirements in this subpart. These written procedures, including any changes, shall be drafted, reviewed, and approved by the appropriate organizational units and reviewed and approved by the quality control unit."
§ 211.160 General requirements.
(a) "The establishment of any specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms required by this subpart, including any change in such specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms, shall be drafted by the appropriate organizational unit and reviewed and approved by the quality control unit. The requirements in this subpart shall be followed and shall be documented at the time of performance. Any deviation from the written specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms shall be recorded and justified."
§ 211.22 Responsibilities of quality control unit.
(a) There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products, and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company.
(b) Adequate laboratory facilities for the testing and approval (or rejection) of components, drug product containers, closures, packaging materials, in-process materials, and drug products shall be available to the quality control unit.
(c) The quality control unit shall have the responsibility for approving or rejecting all procedures or specifications impacting on the identity, strength, quality, and purity of the drug product.
(d) The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed.
Understanding the best practices will help you avoid the common pitfalls and utilize critical thinking throughout the change control process.
One of the top 10 FDA 483 and Warning Letter citations is for inadequate change control. Change control receives detailed scrutiny during FDA inspections, and FDA reviews change control documentation to determine that changes did not adversely impact products, processes, equipment, facilities, etc. A single inadequate change may lead to significant negative events, including release of sub-standard product or product recall. A pattern of inadequate changes may require costly and time-consuming system remediation efforts.
It is therefore critically important to assure that changes are properly described, justified, assessed for risk, implemented, and documented. Changes must also be prospectively reviewed by appropriate subject matter experts. Furthermore, certain major changes (e.g., manufacturing, specifications) may require regulatory filings and/or prior regulatory approval.
To gain a detailed understanding of the above three new aspects and best practices of change control, attend the seminar 'Change Control Best Practices - Avoiding Unintended Consequences of Changes.' This is a practical how-to course is designed to provide participants with skills they can immediately apply to change controls within their own organizations.




