Strategy Planning for Contamination Control in the Manufacture of Biologic Products
To be a winner you need a game plan. To ensure that your organization manufactures winning biologic products with consistent quality, you need to develop a contamination control strategy. The manufacture of biologic products involves complex processes and the use of living cells. These processes and products are vulnerable to contamination by adventitious agents like bacteria, viruses, and fungi. Such microbial contamination can have a huge impact on the manufacturing of biologic products.

- Microbial contamination can lead to product variability
- It can cause changes in impurity profiles
- The levels of bacterial endotoxins can increase
- The degradation or modification of product by microbial enzymes can lead to loss of potency
- Essential drugs can be in short supply when there lengthy shutdown periods, and delays in manufacturing operations during the process of investigations of microbial contaminations.
The root cause for microbial contamination must be identified, and risks associated with them must be controlled in the biologic product manufacture. Contamination must be proactively prevented. A documented contamination control strategy is a key part of good contamination control practices.
However, before developing a documented strategy, you need to consider the key components of a microbial control strategy. This article helps.
- The root cause for contamination
- Regulations and guidance
- 21CFRB'211.28 - provides guidance on the use of protective apparel
- 21CFRB'211.42 - provides guidance for the proper facility design and placement of equipment
- 21CFRB'211.67 - equipment cleaning, sterilization, and maintenance
- 21CFRB'211.100 - production and process controls
- 21CFR parts 600-610 - applicable regulations for therapeutic recombinant products and monoclonal antibodies
- Points to consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use published in February 1997,
- Guidance on Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin (Q5A),
- Guidance on Quality of Biotechnological/Biological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products (Q5D),
- Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products (Q6B)
- Total Organic Carbon Analysis for Cleaning Validation in Pharmaceutical Manufacturing
- Guideline to Aseptic Technique and Clean Room Behavior
- How to Achieve a Compliant and Effective Cleanroom Design and Facility Validation
- Performing an Effective, Robust and Compliant Sterility Failure Investigation: How to Avoid Common Mistakes
- Gaining and Re-establishing Control of Your Cleanroom
- More programs here
- A risk-based approach
- The type of product to be manufactured
- The duration of the product exposure during production
- Whether the facility is used for dedicated or multiple products
- Technologies such as Restricted Access Barrier Systems (RABS) or Isolators provide the required Grade A environment for the protection of the aseptically filled sterile product against contamination from personnel and the surrounding environment. Implementation of such technologies must be considered as part of the process design.
- Measures to control contamination must be designed into each part of the production process. These measures include the use of contamination controls such as cleaning, decontamination, sterilization and transfer methods for primary packaging materials, consumables and intermediate products.
- The validation requirements should be detailed in the validation master plan. This includes validation requirements for the entire cleaning, decontamination, depyrogenation and sterilization processes used all through the process of production
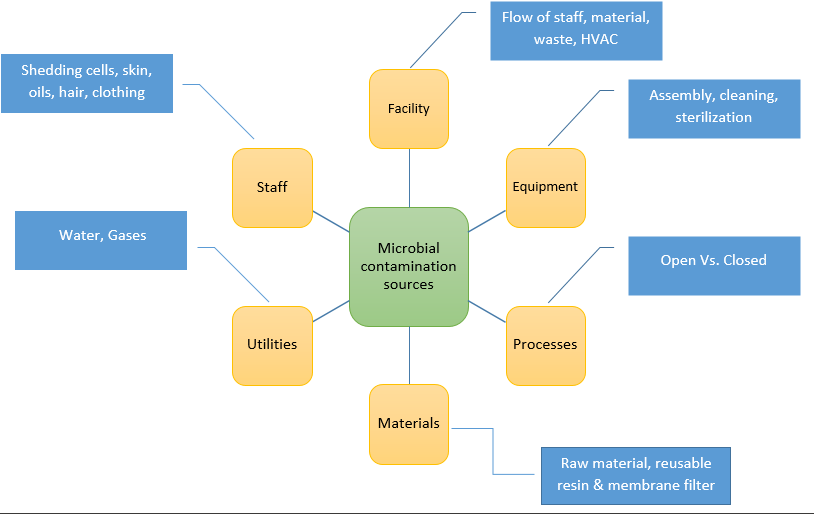
The sources of microbial contamination are illustrated in the following figure.
21CFRB'211 provides the Current good manufacturing practice (CGMP) minimum requirements for preparation of finished human drug products.
The guidance on CGMP for active pharmaceutical ingredients, Q7A
Additional documents
Bite-sized training programs
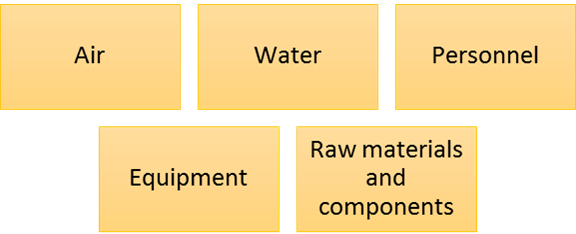
Before developing the strategy, a comprehensive risk assessment must be performed for all possible entry points. This calls for a good understanding of the manufacturing process and the product attributes.
Entry points for Microbes into the cleanroom or other controlled environments
Facility design
Cleanroom design, certification & classification are crucial to contamination control. It should be aligned with the regulatory guidelines. When guidelines do not provide the specifics, the classification of each room should be chosen using a risk-based approach by taking into consideration the following:
The flow of staff, equipment and waste must be designed for effective contamination control. The pressure profile, the air velocity, and air change rates of the cleanroom should be conducive to the quick recovery post a potential contamination event. There should be free movement of airflow from higher to lower classification areas.
It should be ensured that the Utilities which supply the manufacturing process with Purified Water (PW), Water for Injection (WFI), Clean Steam and compressed gases do not contribute to the contamination of the process, environment, or to the finished product. This should be planned as part of the process design.
Process design
Strategies to protect the product from microbial, pyrogen, chemical, non-viable particulate and cross product contamination must be in place during the process design phase.
Personnel
Personnel training is a key component that should be included in the strategy because they are a primary source of contamination. The strategy should include pointers for the practicality, frequency, and continuity of training program including theoretical, practical and cGMP aspects, good gowning techniques, basic microbiology, personal hygiene, aseptic techniques, personnel movement, behavior etc.
Cleaning and disinfection
The maintenance and cleaning details, the applications and procedure details must find an important place in the strategy planning phase.
Environmental monitoring
The monitoring of the environment and the determination of the type and level of microbial and non-viable particulate contamination in a facility is done by a program called the environmental monitoring (EM) program. It provides data that helps in evaluating the adequacy of contamination controls in the cleanroom or manufacturing area.
Process simulation
The performance of the process simulations must be a key element of the contamination control strategy. Process simulation plays an important part in sterile manufacturing. The 'process simulation, also known as a media fill, normally includes exposing the microbiological growth medium to product contact surfaces of equipment, container closure systems, critical environments, and process manipulations to closely simulate the same exposure that the product itself will undergo.' FDA
The initial validation of a facility, product or process begins with process simulations. Thereafter the monitoring of the facility and the monitoring of contamination control is routinely performed through process simulations. They are also performed when there is a significant change to the facility or process.
This article provides you the key elements of a contamination control strategy which you can apply in the strategy planning. You can also find out more about Biotechnology Contamination Control Regulatory Compliance at our bite-sized training sessions.




