Complaints can come from anywhere. Although they cannot be fully eliminated, they can be greatly minimized. If they can't be stopped, they can be efficiently handled. And this requires a good comprehension of the complaint handling fundamentals. This article will help you comprehend and effectively handle them from the opening through the closing of the complaint.
In the medical device industry, complaint handling involves much more than soothing dissatisfied customers. It is a regulatory requirement and a risk-reduction essential.

Understanding Complaints
FDA 820.3
"Complaint means any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution."
ISO 13485:2016
"Written, electronic or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, usability, safety or performance of a medical device that has been released from the organization's control or related to a service that affects the performance of such medical devices."
Complaints are a one-way process. They come in and hence handling them is a reactive process rather than a proactive process. Those involved in the complaint handling process will find reading the 21 CFR Part 820 and ISO 13485:2016 very useful. Also, this seminar sheds valuable insights about the topic.
Differentiating between Complaint and Feedback
A complaint can be feedback. But feedback cannot be a complaint. It is important to understand the difference between feedback and a complaint because you need to set a process for complaints to meet your requirements. You will understand more about it in the latter part of this article. Inquiries about the product, call for preventive maintenance, shipping error, and cosmetic defects are not complaints.
Turning Complaints into Opportunities
Rather than viewing the complaint-handling process as a burden, leading medical device companies highly value them. The data complaints generate helps them develop better, safer products, lower labor costs, and reduce plant downtime. A well-designed complaint capture system is one of the most valued means to monitor the quality of the product and provide feedback to the design, manufacturing, technical support, sales, and marketing teams. Such a system holds a key part of a complete responsive customer service solution and is an important component of risk management.
Sources of Complaints
Complaints can come from various sources and may be submitted by customers or from customer service staff, your sales team, or frontline representatives.
- Telephone
- Online forms
- Email
- Social media
- Online product reviews
- Letters
Whatever the source of the complaint may be, it is important for complaint handling staff to be well equipped to handle complaints in accord with the regulatory requirements. (21 CFR Part 803).
Classifying Complaints
The post-market complaints can be classified into two baskets:
- Incident-driven: An incident such as serious harm or death occurred due to malfunction of the device will require immediate attention.
- Review-driven: When reviewing your data, if a pattern of repeated failures or flaws which qualifies as a complaint are noticed, it can be categorized as a review driven complaint.
Setting up a Process to handle complaints effectively
- FDA 21 CFR 820.198 and ISO 13485:2016 section 8.2.2 provides regulatory requirements about medical device complaint handling.
- As per the requirements, you must have a formally designated unit with clear procedures to receive, review, and evaluate complaints
- They must be investigated in a consistent and timely manner
- You must evaluate whether a complaint warrants reporting to regulatory bodies under FDA 21 CFR Part 803 and/or ISO 13485:2016 section 8.2.3 requirements.
- Your process for complaint management is something that auditors look for when you are inspected.
- Warning letters will be issued if complaint procedures are not in place. Your complaint handling process must be properly documented.
How the FDA expects you to develop and implement proper handling of complaints reportable or non-reportable, product complaint handling and documentation is, is covered in detail here.
A typical Complaint handling process example
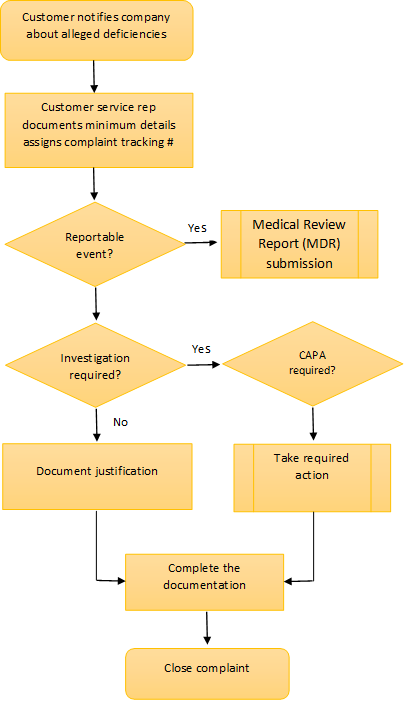
Investigating the complaint
Step 1: Evaluate whether the complaint is valid or potentially reportable
Section 820.198(b) discusses the initial review and evaluation of the complaints in order to determine if the complaints are "valid." this evaluation is not the same as a complaint investigation. The evaluation is performed to determine whether the information is truly a complaint or not and...whether the complaint needs to be investigated or not. If the evaluation decision is not to investigate, the justification must be recorded. - QS Preamble, Comment 190
Review, evaluate and investigate any complaint involving the possible failure of a device, labeling, or packaging to meet any of its specifications, unless such investigation has already been performed for a similar complaint and another investigation is not necessary - 21 CFR 820.198(c)
Step 2: Investigate the probable cause
Step 3: Investigate to root cause where possible
Closing the complaint
After taking appropriate actions, your system may let you close the complaint.
At each step of the investigation process, ensure that you document parallel processes such as vigilance reporting, updating of risk management files, and CAPA if relevant. If your investigation came to an end at the first step, you are required to document that further investigation was not required and the reason for ending the investigation. The same holds good at whatever stage the investigation ends.
If your investigation reached the final stage where you prepare a CAPA, your process may let you end the complaint by providing a reference to the continuing of the CAPA work. This is valuable because you may have received more complaints about the same problem before the CAPA are executed.
Related Trainings
Medical Devices - Post Market Surveillance: Product Complaints Management, Medical Device Reporting, Vigilance Reporting, Product Recalls
The agenda of this Seminar are: The Basics of Quality System Regulations and Post Market Surveillance Activities, Product Complaint Management, Medical Device Reporting, Medical Devices European Vigilance Reporting and Medical Device Recalls. This course will provide practical guidance and real life examples that will help you develop and/or optimize your company’s post market surveillance system.
Navigating the Medical Device Post-Market Maze and Challenges during COVID-19 Pandemic - Learn what FDA is really thinking on regulation and guidance documents
This Seminar will have you stop spinning your wheels with nonessential activities and leave you with a comprehensive learning package that only Rita Hoffman, a former FDA CDRH Recall Branch Chief with experience across the device, drug and veterinary industries can provide.
Document the Clinical Evaluation Report (CER) for the EU MDR and CE Mark
This webinar will discuss the Clinical Evaluation Report (CER). To document the clinical evaluation of a medical device and its output, a Clinical Evaluation Report or CER has to be compiled. The CER is an ongoing compilation of the generation, appraisal, and analysis of clinical data related to a device. It is one of the major components of the Technical Documentation File.
Technical Writing for Medical Devices
This technical writing training program will offer attendees an understanding of how the reporting process supports products in the medical device industry. This virtual session will highlight the mandates for documentation set forth by the regulators, such as the FDA, ISO, and other governing bodies. The program will train attendees on effectively reviewing and revising documents and assessing your audience, in effect producing effective written correspondence.
The 510(k) Mod Program, Breakthrough Technologies, and STeP
What are the U.S. FDA's recently expanded, less traditional / modified pathways for novel technologies or safer products to receive clearand or approval.
US FDA Requirements for Medical Products Labeling / UDI
The U.S. FDA has several requirement for the labeling of medical products. The key requirements are in 21 CFR 801.
Technical Writing for the Pharmaceutical, Medical Device and Biotech Industries
This technical writing training program will offer attendees an understanding of how the reporting process supports products in research, development, and in the marketplace. This virtual session will highlight the mandates for documentation set forth by the regulators, such as the FDA, the ISO, and other governing bodies. The program will train attendees on effectively reviewing and revising documents and assessing your audience, in effect producing effective written correspondence.
Supplier Management with the new Medical Device Regulation EU MDR 745/2017
In this webinar, learn how to implement the changes required by the new regulation EU MDR 745/2017 in your current quality management system in a simple and quick way. The information in this course is vital to your passing the Quality Management Audit by your certification company or notified body.
The New US FDA 21 CFR 820, Quality Management System Regulation (QMSR), Medical Device CGMPs
The US FDA, after years of discussing the harmonization of 21 CFR 820, Device CGMPs (QSR), to the ISO 13485, Device QMS, has finally published their QMSR to replace the previous QSR. On February 02, 2024, they published the final rule in the US Federal Register that revises 21 CFR 820 to include ISO 13485:2016 by reference, called the Quality Management System Regulation (QMSR).
US FDA Medical Device QSR (21 CFR 820) / Quality Management System - current and proposed major changes
The US FDA/CDRH is proposing to harmonize the current Quality System Regulation (QSR) for medical devices (21 CFR 820) by converging its requirements with requirements used by many other regulatory authorities. ISO 13485, into a new device Quality Management System Regulation (QMSR).
Medical Device Cybersecurity Risk Management Training
This webinar will give you a clear structured overview and introduction, into the cybersecurity risk management in relation to the cybersecurity regulation in EU and US and its requirements in the EU and US.
US FDA's AI Framework for Medical Devices
The ability of artificial intelligence / machine learning software to learn from real-world feedback and improve its performance is spurring innovation and leading to the development of novel medical devices. The FDA recognizes AI's advantages and dangers and has started to outline its expectations of industry and device design, programming, unique validation issues, documentation and submissions.
Medical Device Change(s) and the 510(k)
This 90-minute webinar will provide valuable assistance to all regulated companies performing and documenting meaningful, results driven 510(k) / change analysis activities, based on the FDA's new Draft Guidance, "510(k) Device Modifications: Deciding When to Submit a 510(k) for a Change to an Existing Device", dated October 25, 2017.
Risk-based Design Control - The New Paradigm for Medical Device Design
In this webinar, learn about the 2019 approach to compliant design control, how to create, manage, and maintain the information found in the various Risk Management documents and files. Get insights about how to shrink documentation time to provide enough time for actual risk management and mitigation.
US FDA Medical Device QSR, 21 CFR 820 and Quality Management System
Learn the basic overview of the original medical device Quality Management System -- The US FDA’s CGMP, Quality System Regulation (QSR) under 21 CFR 820. Implementation, training requirements and content, and annual internal audit / inspection expectations. The FDA’s four key areas of compliance under QSIT.
Introduction to Medical Device Quality System Regulations
In this webinar, you will get an overview of FDA’s medical device Quality System Regulation, 21 CFR Part 820.
Complaint Management: Best Practices to Assure Regulatory Compliance and Customer Retention
This session will include the requirements for all of the complaint handling responsibilities, which will include defining, documenting, and implementing a complaint-handling system, the requirements for complaint review, investigation, and corrective action, as well as ISO-specific implications. Also covered will be a discussion of what constitutes a complaint, and recommended practice on how to handle "non-complaint" feedback. Also covered will be the application of risk management to a complaint handling system, and a specific risk management system explained.
Mobile Apps as Medical Devices
The rapid expansion and broad applicability of software functions / applications deployed on mobile or other general-purpose computing platforms has created concerns with industry and the US FDA. This has resulted in the FDA focusing on the subset of software functions dealing with medical issues which the Agency intends to apply its authority. What are these issues / software app claims, and how will this affect the medical device industry.
EU MDR 2017/745 Medical Device Classification
In this webinar, you will learn how to classify medical devices according to EU MDR 2017/745 Annex VIII for full compliance with new EU Medical Device Regulation mandatory starting 26-May-2021
Spotlight on Complaint Handling and Medical Device Reporting
This webinar will help you to understand the expectations of complaint handling and medical device reporting. You’ll also learn about lessons from 483s and warning letters. We’ll discuss how you can develop your processes to efficiently and effectively manage complaints and MDRs. Also, we’ll discuss how to link these processes to failure investigation, recalls, and CAPA (Corrective and Preventive Action). We’ll cover best practices, so you can be prepared for an FDA inspection.
Complaint Handling Requirements (US); Interrelationship with CAPA, Change Control, Adverse Event Reporting, Recalls and Life Cycle Process Activities
This training program will provide the regulatory requirements (US) for complaint handling in the medical device and pharmaceutical industries. The course will touch on complaint sources, and details will be furnished on the interrelationships regarding complaint handling/change control/adverse event reporting/and recalls. The course will also include timeline requirements associated to adverse event reporting, which reporting formats to use, when to recall and types of recalls.
FDA Internal Complaint Handling
The information obtained will enable preparation of effective internal systems for receiving, investigating and responding to product complaints during both the investigational and commercial stages. The webinar will address aspects of the review process, SOP preparation, investigation documentation, and response to complainants.
Medical Device Software per IEC 62304
You may find out after seven or more months of waiting that the 510K has been denied because the software portion of the submittal is inadequate and not compliant. Don’t let that happen. This webinar will teach you how to submit compliant and adequate software enabled medical Devices or Software as a Medical Device to the FDA for 510K approval. Also, learn how to avoid the potential audit risk which might prevent you from selling the approved product if you have been found to not have an IEC62304 Compliant Software Quality System.
Scrutinizing Test Method Validation (TMV) to Verify the Performance of a Medical Device
This webinar will help you better understand test method validations to verify the performance of a Medical Device, global reference standards, the FDA requirements and how to perform successful TMV to ensure your inspection of verification is effective, using detailed real-life case studies.
Secure Medical Device Procurement
This Medical Device Security webinar will focus on industry leading practices to assist healthcare delivery organizations (HDOs) with the integration of security into medical device procurement to inform procurement decisions, contractual requirements, and compensating controls. Also attendees will learn the required steps involved in the procurement process, some of which include vendor-level and product-level assessments, contractual requirements, and publicly available resources to assist with standardization.
Complaint Handling, MDR's & Recalls
Medical device firms need to be aware of the benefits of compliance as well as the dangers of inadequate complaint handling. This Webinar will explain the regulatory process and in understandable terms.
EU ISO 13485:2016 Medical Device Quality Management System
Learn the basic overview of the international medical device Quality Management System -- EU’s Quality Management System under ISO 13485:2016. Implementation, training requirements, content and annual internal audit / inspection expectations.
Medical Device Registration - Brazil and Argentina
Learn how to design regulatory strategy for your medical device to expand your business in Brazil & Argentina. Get insight in to regulatory policy and implication for medical device and the key issues in medical device laws of Brazil and Argentina.
Medical Device Employee Training - Requirements and Implementation Tips
This webinar will review types of training requirements from the FDA and ISO 13485 for medical device employee. It will discuss techniques for monitoring and documenting training effectiveness.
Classifying Medical Devices - US and EU
The Medical Device Classification webinar explains the classification system in US and the EU.
Medical Device Software Verification and Validation
This webinar will teach you how to design, build and test medical device software in preparation for the successful 510k submissions. It will cover medical device software user requirements, software architecture, design, unit testing and more.
Medical Device Tracking Requirements
Medical Device Tracking is a very important aspect of the distribution chain of a medical device. This is because it is critical to have a mechanism by which a medical device can be traced and located in its place after it leaves the manufacturer. This system of tracking helps to identify a defective device and ensure that corrective measures are taken immediately.
Medical Device Complaints Handling Strategies for Managing Your Biggest Compliance Challenge
This Medical device complaints handling webinar will discuss the best practices of complaint handling and how to include it in your CAPA program and how to apply risk management to the complaint handling system.
FDA's Medical Device Software Regulation
This training program will focus on FDA software guidance. It will elaborate on applying current provisions that NIST (National Institute of Science and Technology) has put forth in recent reports that FDA will integrate into its regulatory oversight as well.
The Tools the HR Executive Needs to Build an Internal Complaint System that Gets Results!
This webinar will help HR executives design, establish, and maintain an effective system for handling employee complaints in the workplace based on proven best practices that gets results.
Medical Device Complaints, MDR's and Recalls
All medical device firms need to be aware of the benefits of compliance as well as the dangers of inadequate complaint handling. Many medical device firms face regulatory issues as a result of inadequate complaint handling.
Process Validation for Medical Device Manufacturers
This webinar for medical device manufacturers will discuss the CDRH interpretation of the GHTF guidance and how manufacturers should develop process validation plans and conduct production process validation.
FDA Guidance: Medical Device Recalls
This webinar will discuss in detail the FDA guidance on medical device recalls including its role, expectations and enforcement policy. It will cover key topics on medical device recall including recall classification, firm’s responsibilities for a recall, recall communication, recall strategy, FDA expectations and much more.
Medical Device Excise Tax
This webinar explains the excise tax on medical device manufactures that went into effect on January 1, 2013, including the devices it covers and the effective selling price for tax purposes.
Medical Device Reporting for Manufacturers, Importers and Medical Device User Facilities
This webinar on Medical Device Reporting (MDR) will cover FDA requirements for importers, manufacturers and hospitals to report medical device adverse events. It will discuss current policies and guidances for Medical Device Reporting and provide update on upcoming regulations.
Medical Device: Risk Assessment and Mitigation through FMEA
This 90-minute presentation will discuss a structured way to mitigate risks with minimum cost. You will learn how to use FMEA to effectively estimate and assess risks in your medical device.




