Software verification and validation requirements for medical device and their implementation strategies
Your software system must meet the regulatory requirements and be beneficial to the real world. To this end, the FDA requires your system to be validated. The verification and validation (V&V) has become a critical part of the software development process for a medical device. The Requirements traceability matrix (RTM) is a key piece that establishes the system is fully implemented. The RTM documents each of the requirement tracing it to design, items, code, unit, integration and software system test. It is a simple and effective way for documenting your implementation in accord with the expectations of the FDA.

As a best practice, the V&V should be considered early in the design stage when developing requirement specifications for a product. Such an approach will help streamline the overall manufacturing and approval process.
Essential steps in product development process for V& V
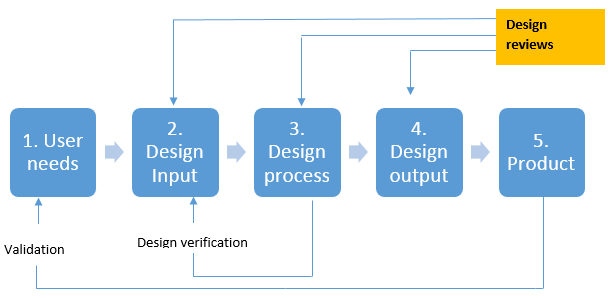
FDA and IEEE definitions for validation and verification
"Software verification provides objective evidence that the design outputs of a particular phase of the software development life cycle meet all of the specified requirements for that phase. Software verification looks for consistency, completeness, and correctness of the software and its supporting documentation, as it is being developed, and provides support for a subsequent conclusion that software is validated. Software testing is one of many verification activities intended to confirm that software development output meets its input requirements. Other verification activities include various static and dynamic analyses, code and document inspections, walkthroughs, and other techniques."
Software validation is the "confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled."
Validation (IEEE) "Confirmation by examination and provisions of objective evidence that the particular requirements for a specific intended use are fulfilled."
Verification (IEEE) "Confirmation by examination and provisions of objective evidence that specified requirements have been fulfilled."
Regulatory requirements
21 CFR B'820.30 Unless specifically exempted in a classification regulation, any medical device software product developed after June 1, 1997, regardless of its device class, is subject to applicable design control provisions.
21 CFR B'820.30 Design controls such as planning, input, verification, and reviews, are required for medical device software.
21 CFR B'820.30(g) Design validation: Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF.
21 CFR B'820.70(i) Any software used to automate any part of the device production process or any part of the quality system must be validated for its intended use. This requirement applies to any software used to automate device design, testing, component acceptance, manufacturing, labeling, packaging, distribution, complaint handling, or to automate any other aspect of the quality system.
21 CFR B'11.10(a). Computer systems used to create, modify, and maintain electronic records and to manage electronic signatures are also subject to the validation requirements. Such computer systems must be validated to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records.
Levels of verification
There are 3 levels of software verification
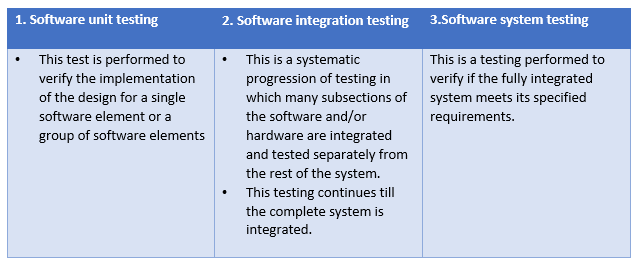
Types of verification
There are 4 types of verification to the 3 levels specified above:

Implementation strategies
- Develop a validation plan (Master plan development) including identifying and analyzing the scope, approach, resources, schedules, the types and extent of the activities, tasks and work items and the approval process. Incorporate compliance with regulatory requirements into the plan.
- Develop an integrated and comprehensive risk analysis taking into consideration the intended use and secondary influencing factors such as interfacing components of the final system/ product or mode of assembly
- Incorporate Test procedures including documentation of input, expected output, actual output, acceptance criteria, information regarding test results, and the test conclusions
- Test execution should be in accord with the test procedures, standard regulatory requirements, and should be documented to ensure compliance with validation regulations
- Process validation to include Installation qualification (I.Q) Operational qualification (O.Q) , and performance qualification (P.Q).
Attend the webinar drafting a software verification and validation report package and protocol to take a deep dive into the documentation required by the U.S. FDA for the verification and validation planning and execution of software after basic developmental testing and de-bug. In this webinar a suggested field-tested 11-element FDA model will be evaluated, implemented, with V&V documentation and test case examples. The focus of the webcast is on the most recent issues the FDA has had in this area, and remediation approaches. Software considered: 1) In-product, 2) As-product, 3) Production and test, and 4) QMS / 21 CFR Pt. 11.
The instructor John E. Lincoln has over 33 years' experience in U.S. FDA-regulated industries and 20 years as a full-time consultant. He has worked with companies from start-up to Fortune 100, in the U.S., Mexico, Canada, France, Germany, Sweden, China and Taiwan.




