The table shows problems or controversies are associated with noncompliance with GMP, inadequate clinical trials and faulty trails which resulted into producing counterfeit or faulty products.
GMP Regulations Set by the United States Food and Drug Administration (FDA), GMP is a standard which stands for Good Manufacturing Practices.
Importance of Following GMP Adhering to GMP regulations helps the production processes as well as the products to be effective, safe, and pure for the consumers. GMP regulations also require that the manufacturers take care to avoid contamination, errors, and mix-ups. The goal of following GMP regulations is to produce a food product or drug that the customer will want to buy and is not dangerous.
Impact of Failure to comply with GMP
- Regulatory Action= warning letters, consent decrees, injunctions, recalls, severe fines
- Negative Exposure= your problems are posted on the FDA website for the whole world to see
- Failure of the “Quality System” = poor quality
- Poor Quality = recalls, unhappy customers
- Unhappy Customers = lost business
- Lost Business = loss of jobs
How to Comply with GMP
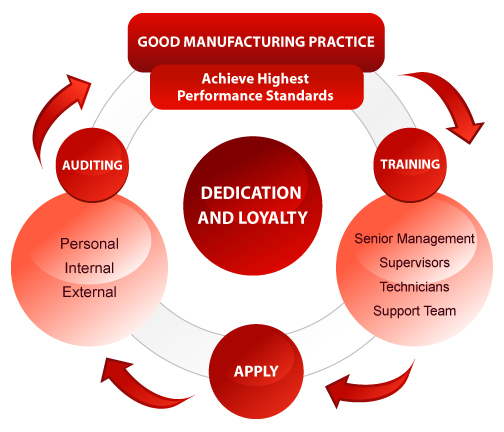 |
The diagram given above illustrates the approach creating and maintaining a GMP lifestyle in a company.
In most of the companies, GMP requirements are very general and open-ended, thereby keeping space for the manufacturer to decide individually to find out the necessary controls.
Steps which should generally be followed for successful implementation and best result of GMPs are
First, decide standards of performance. In these standards, include GMP regulations and other standards which are necessary for the company. Then, train all departments in the company on GMP and other standards. Training should include top management, managers and supervisors, operators and technicians, and support team as they build the most critical and the most important element of successful GMP implementation.
In next stage, apply what is learnt in the training. Responsibility of application of what is learnt lies of the shoulder of the managers and supervisors in a plant. Therefore, it is important that managers and supervisors be involved in training, so that they can support it through application.
The third stage is auditing as only auditing can ensure your efforts have provided adequate controls and fruitful results of training.
Finally, analyze the results of audits to know whether your standards of performance are needed to be modified.
However, dedication and loyalty towards good work and company is the key to successful implementation and use of GMP in every organization. And this dedication is required from all levels of the organization. Train your employees, fuel their commitment and experience how GMP becomes the lifestyle of your company.
Clinical Trails
Clinical trials are a critical part of the research process involved in producing a drug.
Importance of Clinical Trails
Clinical trials contribute to knowledge of and progress in the fight against the disease and at the same time provides crystal-clear understanding of the success (or failure) of the medicine. Many of today's treatments are based on what we learned from clinical trials in the past. Greater participation in clinical trials means faster answers to critical research questions which lead to better treatment for all diseases.
Securing Safety of the Participants in Clinical Trials
People involved in the clinical trial process are required to secure the safety of the participants of the clinical trial. Sponsors, local site investigators, IRBs supervising the trials, and often the regulatory bodies of the country or region where the drug is being manufactured or will be sold, remain responsible for ensuring the safety of the participants of the clinical trial.
Sponsor
During the clinical trial process, a sponsor remains responsible for –
- Informing the local site investigators accurately of the true past safety record of the drug, device or other medical treatments which is to be tested;
- Monitoring the results coming from different sites;
- Collecting adverse effect news and inform the investigators to find out what went wrong in the trial
Local Site Investigator/Physician
- The investigator physician plays the pivotal role in securing the safety of the subjects taking part in trials. In case he finds the drug unsafe for the participants, he can call off the trial;
- Conducting the trial in accordance with the protocol and supervising the staff doing the trial throughout the process, are the responsibilities of the investigator;
- Reviewing the adverse effect report sent by the sponsor.
IRBs
- Function of the IRB is to scrutinize study protocol, drug history and safety of the participants.
Regulatory Bodies
- In case of new drugs, regulatory bodies review all the study data before allowing the drug to be marketed. Regulatory body such as the FDA can do random audit of files and/or local sites to invigilate procedures of drug manufacturing.
When the FDA Issue Warning Letters
In case of improper monitoring of the clinical investigations [21 CFR 312.50; 312.56(a)]
When company fails to conduct an investigation in accordance with the general investigational plan and protocols specified by the IND [21 CFR 312.50]
In case of failure to ensure that investigators who were qualified by training and experience, were selected as appropriate experts to investigate a drug [21 CFR 312.53(a)]
In case of improper preparation and maintenance of adequate and accurate data, case histories relevant for the trial, FDA can issue warning letter [21 CFR 312.62(b)].
Failure to preserve adequate records of the disposition of the drug, including dates, quantity, and use by subjects can also call for warning letter [21 CFR 312.62(a)].
In case the informed consent is not obtained in accordance with [21 CFR Part 50].
Key Regulations and Guidance Documents
- 21 CFR Part 11 Electronic Records: Electronic Signatures
- 21 CFR Part 50 Protection of Human Subjects
- 21 CFR Part 54 Financial Disclosure by CIs
- 21 CFR Part 56 Institutional Review Boards
- 21 CFR Part 312 IND
- 21 CFR Part 314 NDA
- ICH-E6 GCP: Consolidated Guidance
How to Conduct Clinical Trial For
pharmaceutical development, steps of clinical trials are:
- Pre-clinical testing in animals
- Submission of IND application
- Clinical Trials in humans: Phase I, Phase II, and Phase III
- Submission of new drug application
- Product sales and marketing – Phase IV
Steps for Pre-Clinical Development
- Discovering Drug
- Where possible, assess efficacy of the drug
- Establish pharmacokinetics and pharmacodynamics (ADME)
- Work on developing analytical and chemical methods (quality control, blood levels)
- Evaluate safety of the drug and to do that perform toxicology studies
- Formulate procedures for clinical trials
- Work on manufacturing process
- Find out product stability
According to FDA, any good clinical trial should follow the basic principles of good clinical practices (GCPs) and human subject protection (HSP). Principles of GCP are regarded as law or regulation in many countries. In FDA's regulations for conducting clinical trials, both GCP and HSP have been addressed prominently.
Clinical Trial and Good Clinical Practices (GCP)
Good Clinical Practice or GCP is an international quality standard provided by ICH (International Conference on Harmonization). GCP guidelines ensure safety and efficacy of a newly developed drug and also protect human rights in a clinical trial.
GCP provides a crystal clear understanding of how clinical trials should be conducted and also defines the roles and responsibilities of the clinical trial sponsor, investigator and monitors.
Drug Labeling
Regulated by the Food and Drug Administration's Division of Drug Marketing, Advertising, and Communications, drug labeling refers to all of the printed information that comes along with a drug, including the label, the wrapping and the package insert.
Drug labeling regulations apply to prescription drugs, over-the-counter (nonprescription) drugs, and dietary supplements. Training
FDA Rules of Labeling for NDA and OTC Drugs
For NDA and OTC drugs, FDA has some stipulated norms to which differentiate both these drugs:
| |
FDA for NDA Drugs
|
|
|
|
|
| |
|
FDA pre approval for NDA drugs is mandatory
|
|
|
|
|
For OTC drugs, FDA pre approval is not mandatory
|
|
|
| |
|
Clinical studies are also mandatory for NDA drugs and the studies involve user fees
|
|
|
|
|
Clinical studies are not mandatory and do not involve user fees
|
|
|
| |
|
FDA approval for each NDA drug is obligatory, FDA must approved labeling that is unique to each specific (prescription) NDA drug
|
|
|
|
|
FDA approval for labeling of OTC drugs is not obligatory, same labeling for all similar drugs that meet OTC Monograph in CFR
|
|
|
| |
Marketing of drugs can be exclusive
|
|
|
No exclusivity is granted for marketing OTC drugs
|
|
| |
|
FDA approval license the NDA drugs to market in US
|
|
|
|
|
Final Monograph is open to anyone
|
|
|